
More than 30 years ago, Congress overwhelmingly passed a landmark health bill aimed at motivating pharmaceutical companies to develop new drugs for people whose rare diseases had been ignored.
By the drugmakers’ calculations, the markets for such diseases weren’t big enough to bother with.
But lucrative financial incentives created by the Orphan Drug Act signed into law by President Ronald Reagan in 1983 succeeded far beyond anyone’s expectations. More than 200 companies have brought almost 450 “orphan drugs” to market since the law took effect.
Yet a Kaiser Health News investigation shows that the system intended to help desperate patients is being manipulated by drugmakers to maximize profits and to protect niche markets for medicines already being taken by millions. The companies aren’t breaking the law but they are using the Orphan Drug Act to their advantage in ways that its architects say they didn’t foresee or intend. Today, many orphan medicines, originally developed to treat diseases affecting fewer than 200,000 people, come with astronomical price tags.
And many drugs that now have orphan status aren’t entirely new. More than 70 were drugs first approved by the Food and Drug Administration for mass market use. These medicines, some with familiar brand names, were later approved as orphans. In each case, their manufacturers received millions of dollars in government incentives plus seven years of exclusive rights to treat that rare disease, or a monopoly.
Drugmakers of popular mass market drugs later sought and received orphan status for the cholesterol blockbuster Crestor, Abilify for psychiatric conditions, cancer drug Herceptin, and rheumatoid arthritis drug Humira, the best-selling medicine in the world.
More than 80 other orphans won FDA approval for more than one rare disease, and in some cases, multiple rare diseases. For each additional approval, the drugmaker qualified for a fresh batch of incentives. Botox, stocked in most dermatologists’ offices, started out as a drug to treat painful muscle spasms of the eye and now has three orphan drug approvals. It’s also approved as a mass market drug to treat a variety of ailments, including chronic migraines and wrinkles.
Altogether, KHN’s investigation found that about a third of orphan approvals by the FDA since the program began have been either for repurposed mass market drugs or drugs that received multiple orphan approvals.
“What we are seeing is a system that was created with good intent being hijacked,” said Bernard Munos, a former corporate strategy advisor at drug giant Eli Lilly and Co. who reviewed the KHN analysis of several FDA drug databases. It’s “quite remarkable that it has gone on for so long.”
And the proportion of new drugs approved as orphans has ballooned. In 2015, 21 orphan drugs were approved, accounting for 47 percent of all new medicines, up from just 29 percent in 2010; in 2016, nine more orphans won approval, 40 percent of the total.
When a drugmaker wins approval of a medicine for an orphan disease, the company gets seven years of exclusive rights to the marketplace, which means the FDA won’t approve another version to treat that rare disease for seven years, even if the brand name company’s patent has run out. The exclusivity is compensation for developing a drug designed for a small number of patients whose total sales weren’t expected to be that profitable.
But the exclusivity is a potent pricing tool. Drugmakers can charge whatever they want by shielding their medicine from competition. The market exclusivity granted by the Orphan Drug Act can be a vital part of the protective shield that companies create. What’s more, manufacturers can return to the FDA with the same drug again and again, each time testing the drug against a new rare disease.
Critics have assailed drugmakers in the past for gaming the orphan drug approval process. But the extent to which companies have been winning approval for drugs that aren’t what advocates call “true orphans” hadn’t been documented until the Kaiser Health News investigation.

Bernard Munos, senior fellow at the Milken Institute (Courtesy of Bernard Munos)
Munos said he was “shocked” by the sheer number of mass market drugs being repurposed as well as those approved multiple times.
Even agency officials said they weren’t aware of the scope of the issue. After reviewing KHN’s findings for two weeks, Dr. Gayatri Rao director of the FDA’s Office of Orphan Products Development said she “appreciated the work” and expressed interest in studying how often drug companies are “repurposing” a drug for a new rare disease, or taking “multiple bites of the apple.”
“We are going to look into this,” she said, adding that she could consider a regulatory change.
Rao pointed out that the “repurposing” of drugs does have scientific and patient benefits. For example, cancer drugs approved for one type of malignancy can be tested and approved for others. Gleevec, a drug that revolutionized the treatment of chronic myeloid leukemia, now has nine orphan approvals.
But in a 2015 commentary published in the American Journal of Clinical Oncology, Dr. Martin Makary at the Johns Hopkins University School of Medicine focused on cancer drugs including Gleevec, arguing that the drug was never meant to serve an orphan population. Instead, Makary and his team wrote, drugmakers purposely identify small patient populations to gain additional approvals — a process he described as “salami slicing.”
“By salami slicing the disease into subgroups, it allows them to get the orphan drug approval with all the government benefits and even some of the subsidies,” Makary said. The prices of such medications often rise because they have seven years without competition for a new set of patients, Makary added.
The FDA has taken a different view of repurposing.
“We always talked about how we permit the second bite of the apple, third bite of the apple, as one small way to incentivize repurposing,” Rao said, noting that industry and patient groups have been pressing the FDA for even stronger incentives. “Now, all of sudden, it seems like, wow, this practice may be driving up prices.”
Novartis, which owns Gleevec, said in an email statement that the company is advancing research and following the science to “bring the right treatments to the right patients based on unmet need, not the size of the patient population.”
Two KHN reporters spent six months analyzing data and talking to lawmakers, patients, advocates, doctors and companies to understand how the FDA’s orphan drug program has evolved amid a national uproar over soaring drug prices. President-elect Donald Trump vowed on the campaign trail to bring down prescription drug prices and during a Jan. 11 press conference said the drug industry is “getting away with murder.”
The investigation examined how drugmakers use the law to their advantage — often with the guidance of former FDA officials — and have made the development of medicines that were once thought to be business backwaters into one of the pharmaceutical industry’s hottest sectors.
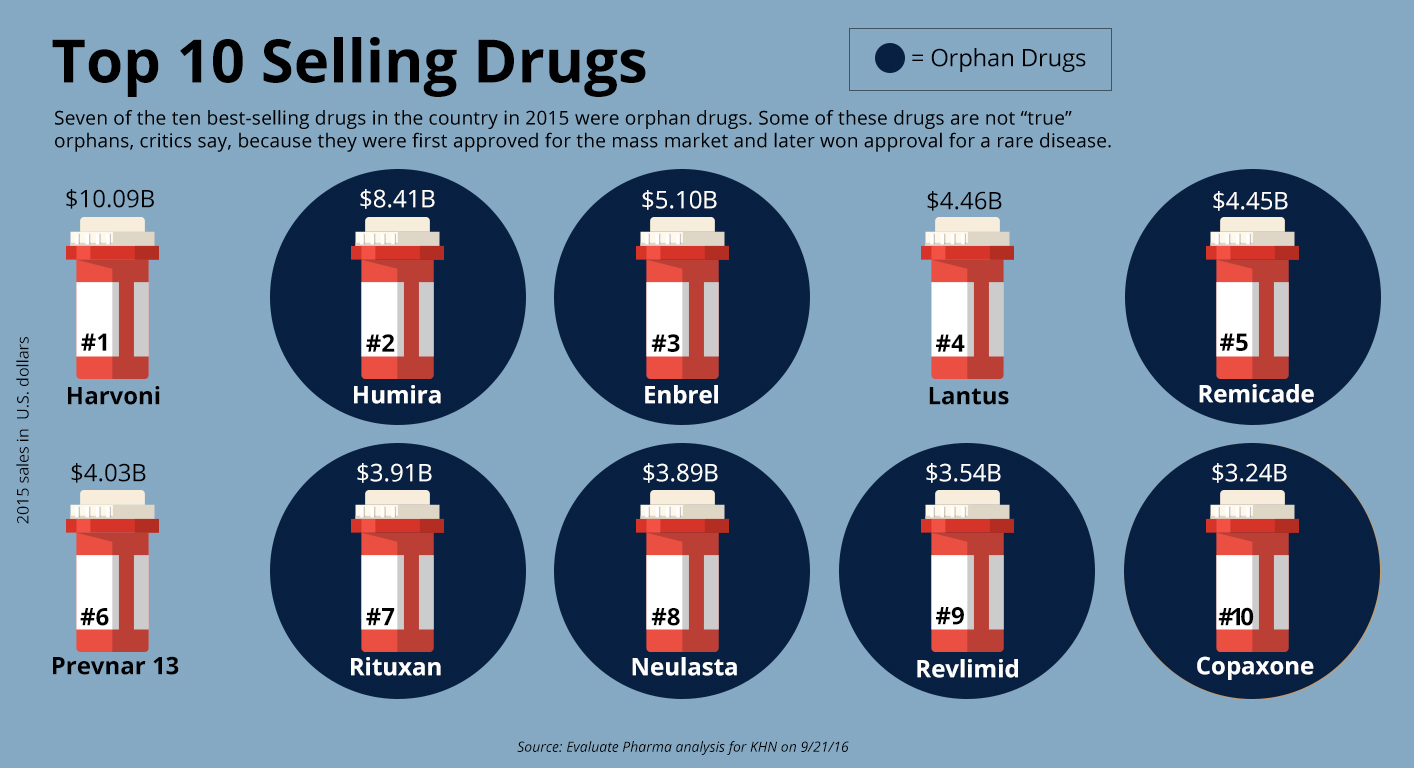
Orphan drugs now account for seven of the 10 top-selling drugs of any kind, ranked by annual sales, according to EvaluatePharma.
“Orphans are wicked hot,” said Dr. Tim Coté, a former FDA official who now runs a consulting firm that advises drugmakers on orphan drugs.
No one disputes that orphan drugs have helped or saved hundreds of thousands of patients suffering from debilitating or even fatal rare diseases. Exactly how many is difficult to estimate because the FDA doesn’t track such information.
And drug industry officials say companies should be rewarded, not punished, for making those treatments possible and for pursuing new drugs that aren’t always an economic success.
Research and development is “long, costly, risky,” said Anne Pritchett, vice president, policy and research at industry lobbying group PhRMA. “When you look at cystic fibrosis it was 25 years to the development of an effective therapy … I think we would be concerned about anything that would undermine the current [orphan drug] incentives.”
Dead children … people are willing to pay a lot to prevent that.
Former U.S. Rep. Henry Waxman, D-Calif., a champion of the 1983 Orphan Drug Act takes a different view.
“The Orphan Drug Act has been a great success because many people with diseases that affect very few people now have drugs available to them,” Waxman said. “But it’s been in some ways turned on its head when it becomes the basis of manipulating the system for the drug company to make much more money than they would in an open, competitive market.”
Booming Business
On a late summer day, Tim Coté sat in a corner office of his Sandy Spring, Md., consulting firm, Coté Orphan. He leaned into his computer microphone to dispense insider knowledge about the orphan drug approval process on a webcast hosted by FDAnews, a trade news organization. Listeners paid about $300 a head, but Coté said he wasn’t paid for doing it.
The FDA is more flexible in evaluating drugs for rare diseases, he said, explaining that “about half of them get through with just one pivotal clinical trial. Not so for common diseases.” The FDA, citing a report from the National Organization for Rare Disorders, said about two-thirds of orphan drugs were approved with one adequate and well-controlled trial with supportive evidence. It typically requires two or three such trials to approve a mass market drug.
Coté also told the webinar audience that clinical trials for orphan drugs are usually smaller and the approval is a “different scientific and regulatory experience.”
Coté knows his stuff. He was Rao’s immediate predecessor as chief of the FDA’s Office of Orphan Products Development. It’s not unusual for government officials to leave FDA and other regulatory agencies and obtain jobs as consultants or industry executives.

Dr. Tim Coté, former head of the FDA’s orphan drug office and now principal and chief executive of Coté Orphan (Courtesy of Coté Orphan)
Coté’s website, headlined “The Inside Track,” notes that he oversaw applications that led to the approval of at least 150 orphan drugs when he was at the FDA and that his firm is now the largest submitter of orphan drug applications.
“We write the entire application,” the website for Coté’s company notes, adding that his staff of 25 includes regulatory scientists with deep knowledge and experience in FDA’s “unwritten rules” regarding orphan drugs.
Many of Coté’s more than 300 clients are small biotech companies begun by researchers or even passionate parents who found investment backing. Parents Ilan and Annie Ganot, for example, started Solid Biosciences to find treatments and potentially a cure for their son with Duchenne muscular dystrophy.
Coté guides them through the regulatory process since most don’t have the expertise. He can offer his expertise and develop an application that makes it easier for the FDA to designate and approve the drug.
“When you make the FDA smile, the value of your asset goes up. And that’s how the game is played,” he said in an interview, adding quickly, “It’s really not a game because people’s lives are what is in balance.”
Coté and other ex-FDA officials play a vital role in helping drugmakers choose rare disease targets and get through the FDA approval process.
Search for drug or disease
Approved Orphan Uses
A small cottage industry has grown around the Orphan Drug Act. Dr. Marlene Haffner, who preceded Coté in the FDA’s orphan office, started her own consulting firm, too, to advise small and large companies on orphan drug applications. A third company is Camargo Pharmaceutical Services, led by industry veterans and former FDA officials, which advises companies focused on repurposing drugs for orphan approval. The firm tries “to be in front of the FDA a lot — three to four times a month,” said Jennifer King, Camargo’s director of marketing. Fees for consulting on orphan drugs industry wide range from $5,000 to $100,000, depending upon what services are provided, Coté said.
Getting through the orphan approval process involves a series of steps.

Dr. Marlene Haffner, former head of FDA’s orphan drug office and founder of Haffner Associates (Courtesy of Marlene Haffner)
First, drugs must be designated by the FDA as potential candidates for approval. A company has to demonstrate that its drug is a promising treatment for a disease that affects fewer than 200,000 patients. If the FDA agrees and makes the formal designation, financial incentives kick in, including a 50 percent tax break on research and development (R&D) and access to federal grants.
When drugs get orphan designation, companies often reap other financial rewards. Shares in publicly traded companies often rise on the news — sometimes soaring as high as 30 percent. That happens, in part, because orphans have a track record of being approved at much higher rates than drugs for common diseases.
The 50 percent R&D tax credit pays off, too. In 2012, one of the biggest orphan drug companies, BioMarin, received $32.6 million from a combination of federal and state of California tax credits. BioMarin spokeswoman Debra Charlesworth confirmed that the orphan credit made up the “vast majority” of that deferred tax benefit. She also noted that credit “has successfully fueled an industry that didn’t previously exist” and led to more rare disease research.
Industry-wide, orphan drug tax credits cost the federal government $1.76 billion in fiscal 2016 — roughly what President Barack Obama asked Congress to spend to fight the Zika virus before a $1.1 billion expenditure was approved. And, because so many orphan drugs are under development, the U.S. could grant nearly $50 billion in tax credits from 2016 to 2025, estimates the Treasury Department.
There’s a lot of creativity behind figuring out how to make a drug an orphan.
In Coté’s webinar and in multiple interviews, he described many ways companies can win orphan status. They can test their drugs on children with adult diseases, such as schizophrenia, or find drugs for ailments like malaria that are uncommon in America.
“African sleeping sickness: horrible problem in Africa but not here, not in the U.S.,” Coté told his webinar audience. “So a drug development effort that was aimed toward some of these tropical diseases can actually get all the benefits of the Orphan Drug Act.”
People have played games with the Orphan Drug Act since it was passed.
Another popular strategy is to create “follow-on drugs” that represent incremental steps forward.
About 30 percent of Coté’s clients are companies looking to improve upon some other orphan drug “which just made billions and billions,” he said in an interview.
Repurposing an already approved drug is another strategy his firm promotes. In a video posted on his website in July, Coté explained the advantages for companies that can move directly into a clinical trial without much preparatory work because the drug’s safety has already been demonstrated.
“All you gotta do is establish that the product can work in this new orphan indication,” he said, adding tips on how to do it and still make money.
‘That Is Not A True Orphan Drug’
Turning mass market drugs into orphans has been a familiar path for some of the most popular drugs ever discovered.
AbbVie’s Humira is the best-selling drug in the world, and most of its sales are in the U.S. where revenue reached $7.6 billion through the third quarter of 2016 and $11.8 billion worldwide, according to the company’s latest financial report.
Humira was approved by the FDA in late 2002 to treat millions of people who suffer from rheumatoid arthritis. Three years later, AbbVie asked the FDA to designate it as an orphan to treat juvenile rheumatoid arthritis, which they told the FDA affects between 30,000 and 50,000 Americans. That pediatric use was approved in 2008, and Humira subsequently was approved for four more rare diseases, including Crohn’s and uveitis, an inflammatory disease affecting the eyes.
The ophthalmologic approval would extend the market exclusivity for Humira for that disease until 2023. When asked why AbbVie sought multiple orphan designations and approvals for Humira, the company declined to comment.
Peter Saltonstall, executive director of the National Organization for Rare Disorders, said that Humira is “not a true orphan drug.” But, he said, the company has “the ability to go out and get orphan designation. That’s the way the law reads right now … they can do whatever they want to do.”
It is difficult to say exactly how or if orphan exclusivity affects the price of Humira, which is a complex biologic drug and also has been protected by numerous patents. The drug has long been AbbVie’s top seller, accounting for 63 percent of its revenues, according to its most recent financial filing.
EvaluatePharma notes in its recent report that Humira, as well as a handful of other top drugs, receive less than 25 percent of their sales from orphan uses. Still, if Humira’s orphan uses accounted for just 10 percent of annual sales, the revenue would surpass $1 billion.
By stacking up a series of orphan disease approvals, one seven-year exclusivity period leads into another, maximizing the length of a company’s monopoly. Sigma-Tau Pharmaceuticals, for example, had some form of orphan exclusivity over its metabolic disorder drug for more than 20 years. The drug, Carnitor, received a second orphan approval four months before its first exclusivity was set to expire. And it won its third orphan approval, for an IV formulation of the drug, just one day before its second exclusivity period was set to expire in December of 1999.
“The sequence and timing of regulatory filings for Carnitor reflect the time required to conduct large controlled clinical trials, as well as evolving medical strategies and regulatory pathways pursued by different sponsors over many years,” said GianFranco Fornasini, senior vice president of scientific affairs at Sigma-Tau.

Dr. Gayatri Rao, current director for the FDA’s Office of Orphan Products Development (Courtesy of FDA)
The FDA’s Rao said each new exclusivity period is disease-specific and once any seven-year period runs out, generics can come in. Gleevec, for example, won FDA approval to treat several kinds of rare cancer. All but one of its orphan exclusivity periods had expired by 2015, allowing two generics to enter the marketplace. But Gleevec still has exclusivity until 2020 to treat newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia in patients who are also on chemotherapy.
It’s also true, Rao explained, that some of the drugs that go through the orphan process may not specifically treat a rare disease. For example, a very toxic cancer drug might may not work well in earlier stages because its risks outweigh the benefits. But the company may propose that it will help a smaller group of later-stage cancer patients and win orphan approval just for that group.
Former FDA orphan drug director Haffner said her FDA office worked on rules defining how companies could legitimately pursue approval for a small group of patients with a specific unmet medical need.
“People have played games with the Orphan Drug Act since it was passed,” said Haffner, who first took a job with drugmaker Amgen after leaving the FDA and then became an independent consultant. “It’s the American way, I don’t mean that in a nasty way. But we take advantage of what’s in front of us.”
In 2013, the FDA clarified the Orphan Drug Act’s regulations and said it wanted to avoid the possibility that some companies could “potentially ‘game’ approvals by seeking successive narrow approvals of a drug.”
In reality, Rao said, the regulations did not really change “much of what our practice was.” The agency wanted to address what Rao said were “common misconceptions” and frequently asked questions so officials changed wording in the regulations to better define exactly what could be considered an orphan drug.
Breaking down larger, broader diseases into smaller groups is still allowed under certain conditions and companies can still win multiple orphan approvals for a single drug — even if the total population served rises above the 200,000 mark.
Amgen Inc.’s Repatha won marketing approval and exclusive rights in 2015 for the orphan disease homozygous familial hypercholesterolemia, which affects a population of about 300 people in the U.S. On the very same day, the drug was approved as a mass market drug to treat up to 11 million people with uncontrolled levels of LDL cholesterol, said Amgen spokeswoman Kristen Davis.
Dr. Steven Nissen of the Cleveland Clinic, who ran a broader trial on Repatha, said, “It’s certainly not considered by any of us to be an orphan drug.”
Safeguarding The ‘Prize’
Considering the long history of what’s happened, Tim Coté acknowledges that there are “some loopholes” in the Orphan Drug Act. Perhaps 3 percent or less of approved orphans were not in the “spirit” of the law, he said.
But Coté, rare disease advocates, patients and people in the drug industry expressed fear that changing the Orphan Drug Act or questioning its success would hurt the development of drugs for rare patients.
Former U.S. Rep. Jim Greenwood, R-Pa., now president of the Biotechnology Innovation Organization, an industry trade group, said that concerns about high prices for orphan drugs aren’t justified. The incentives, he said, should not be altered because rare diseases are “tragically killing and brutalizing mostly children.”
Greenwood seemed unaware that dozens of orphan approvals stemmed from the repurposing of mass market drugs, like Humira or Enbrel, another drug developed first for rheumatoid arthritis. Still, he said, “I would argue that the risk of losing incentives in the system far outweighs the benefit of trying to save a few pennies on the health care dollar.”
It’s a sentiment that Coté and other advocates share. While talking about the $311,000 annual price tag for cystic fibrosis drug Kalydeco, Coté said any parent whose child has the disease would be a big fan of the drug.
“The price point is justified because actually it has a dramatic effect on the children. Dead children … people are willing to pay a lot to prevent that,” Coté said. “And that’s a real good thing that we have this drug. OK?”
The first drug to specifically target the underlying biochemical defect of cystic fibrosis, Kalydeco is approved to treat a subset of patients who have specific mutations in their genes. Development of the drug was financed by the Cystic Fibrosis Foundation, which sold its rights to sales royalties from Kalydeco and other cystic fibrosis drugs for $3.3 billion in 2014.

Others, including Henry Waxman, are far more critical and have tried to do something about it over the years. Waxman proposed multiple bills to rein in corporate profits by amending the orphan drug law that he sponsored, but none succeeded.
The FDA has also tried but failed, to keep corporations in check.
In 2012, drugmaker Depomed Inc. filed suit against the FDA for refusing to give its drug Gralise seven years of market exclusivity as a treatment for pain related to shingles.
Rao said the agency wanted to see proof that Gralise was clinically superior to other drugs, noting there “were a bunch of other generics on the market” with different formulations and dosing requirements. Gralise’s active ingredient, gabapentin, is the same one as in Pfizer’s mass market blockbuster Neurontin, which is also approved for treatment of shingles pain.
The FDA approved Gralise but denied seven years of exclusivity.
In response, the drugmaker sued the agency and won its case, arguing that according to the law, they didn’t have to prove their drug was clinically superior to gain the monopoly.
Today, the drug is one of Depomed’s top products with sales of $81 million in 2015. And, in a recent proxy statement, Depomed listed “protecting Gralise exclusivity” as a corporate objective under the category of “enhance and protect future cash flow.” Its orphan exclusivity ends in 2018. Depomed spokesman Christopher Keenan said Gralise wanted patent exclusivity to block competition. But, Keenan said, “Had the patent effort failed on all fronts, the Orphan Drug Designation would have been very important.”
After reviewing KHN’s analysis, Rao said she wants to better understand why drugmakers are applying for multiple approvals and has asked for a case-by-case review of all orphan designations granted in 2010 and 2015. She said the agency lacks the resources to run an analysis of the entire orphan drug database.
“Our goal is to try to get it right,” she said. “There are over 7,000 rare diseases, likely more, the vast majority of which have nothing … I want to ensure that we continue to keep our eye on that prize.”
Contributors: John Hillkirk, Scott Hensley at NPR, Diane Webber, Marilyn Thompson (editors); Elizabeth Lucas (data editing); Joe Neel at NPR (radio editing)
Interactives, video and presentation: Lydia Zuraw, Emily Kopp, Meredith Rizzo at NPR (digital presentation); Francis Ying (videos, motion graphic); Heidi de Marco (videos, photos, audio); Alley Interactive (database lookup)
KHN’s coverage of prescription drug development, costs and pricing is supported in part by the Laura and John Arnold Foundation.



